

1. Introduction
The use of dye-sensitized semiconductors in photochemical water splitting is currently attracting a lot of attention. In these materials, a visible light-absorbing dye is adsorbed onto a wide-gap semiconductor and a cocatalyst acts as an active site for hydrogen evolution. Upon excitation of the dye by visible light, an electron is transferred to the semiconductor. Electrons then move to the cocatalyst (typically, nanoparticulate Pt), and are used for proton reduction, resulting in H2 evolution. The oxidized dye that has lost an electron is reduced by a solution-phase electron donor. This photoreduction process may be studied as a hydrogen-evolving “half cell” by using a sacrificial electron donor, or as a full water splitting system by using a reversible redox couple such as I3-/I-. Dye-sensitized photocatalysts are highly designable with many choices in the semiconductor and the dye [1⇓⇓⇓⇓⇓⇓⇓⇓⇓-11]. Although it is difficult to oxidize water with the oxidizing power of dyes, Z-scheme water splitting is achievable when the hydrogen evolving photocatalyst is combined with a photocatalyst for oxygen evolution such as WO3 or BiVO4.
Pt-loaded TiO2 is one of the best studied materials as the component for dye-sensitized H2 evolution. While Pt/TiO2 is an active photocatalyst for water splitting [12⇓-14], it works only under UV irradiation because the band gap energy of TiO2 (3.0-3.2 eV, depending on the polymorph) is beyond the range of visible light. A Pt-loaded P25 sample that contains anatase and rutile as the main phases has been shown to have an apparent quantum yield of ~60% for H2 evolution under 300 nm illumination when methanol is used as a sacrificial electron donor [15]. The high quantum yield implies that hole scavenging by methanol is rapid compared to electron-hole recombination, and presumably this is because conduction band electron transport to the Pt co-catalyst is fast. Modification of Pt/TiO2 with a photosensitizer such as a Ru(II) tris(bipyridine) derivative, a phthalocyanine- or coumarin-dye enables this process to occur with visible light excitation when an electron donor is added to the solution [1⇓⇓⇓-5]. The development of dye-sensitized water splitting systems has focused mostly on the refinement of the dye and semiconductor components [10,16]. With numerous efforts made so far, apparent quantum yields of several tens of percent under visible light have been achieved using suitably modified Pt/TiO2 [3]. Visible-light H2 evolution over dye-sensitized Pt/TiO2 using a reversible electron donor (e.g., I-) has been reported as well [2,6,7]. This suggested that dye-sensitized Pt/TiO2 could become a H2-evolving photocatalyst for Z-scheme overall water splitting, in combination with a reversible donor/acceptor pair and an O2-evolving photocatalyst. However, photochemical water splitting in a Z-scheme has not yet been reported using dye-sensitized Pt/TiO2. A plausible explanation for the low quantum yield in this system is the fast kinetics of back electron transfer between Pt/TiO2 and the oxidized donor (i.e., I3-) [2]. In dye-sensitized photocatalysts using a bulk-type semiconductor such as TiO2 and SrTiO3, semiconductor-to-dye back electron transfer has been studied [17⇓⇓⇓⇓⇓-23]. However, the back electron transfer to the oxidized electron donor has not been studied.
In earlier studies, we have found that the back electron transfer reaction to I3- could be suppressed by modifying the semiconductor surface with anionic polymers such as sodium poly(styrenesulfonate) or polymethacrylate (abbreviated as PSS or PMA, respectively), as unveiled by transient absorption spectroscopy [24⇓-26]. Anionic polymer modification has recently been shown to be the key to induce the full potential of dye-sensitized HCa2Nb3O10 (Pt(in)/HCa2Nb3O10) for overall water splitting in a Z-scheme [25,26]. Here one may pose a question: How well does Pt/TiO2 with these surface modifications work in a non-sacrificial system for visible light H2 evolution? Here, we study the effects of surface modification of Pt/TiO2 sensitized by [Ru(4,4′-(CH3)2-bpy)2(4,4'- (PO3H2)2bpy)]2+ (bpy = 2,2'-bipyridine), abbreviated as RuP, on the efficiency of visible light-driven hydrogen evolution. As surface modifiers, we employ not only anionic polymers (e.g., PSS and PMA) but also Al2O3, which we have used to slow down back electron transfer from the semiconductor conduction band to the oxidized sensitizer [20,27]. The results obtained using Pt/TiO2, including the kinetics of back electron transfer to a non-sacrificial electron donor (I3-), are compared to those of Pt(in)/HCa2Nb3O10, which is the most efficient system for Z-scheme water splitting among dye-sensitized photocatalysts [25,26].
2. Experimental
2.1. Synthesis of Pt/TiO2
A photodeposition method was applied to Pt nanoparticle deposition onto TiO2 (AEROXIDE P25; TCI). TiO2 (0.3 g) was suspended in aqueous methanol solution (140 mL, 15 vol%) containing dissolved H2PtCl6⋅6H2O (0.10 wt% Pt vs. TiO2). The suspension was photolyzed with a UV light source (λ > 300 nm) for 1.5 h. After irradiation, the solid was separated by filtration, rinsed with water, and collected as a powder after drying at 343 K. Pt(in)/HCa2Nb3O10 was synthesized as described previously [28].
2.2. Synthesis of RuP/Pt/TiO2
RuP was prepared as the dichloride salt as previously described [29]. The structure and purity of RuP was established by 1H NMR and UV-vis absorption spectroscopy. RuP was adsorbed by a stirring method [30]. Pt/TiO2 powder was suspended in aqueous RuP solution, whose pH was set to be 2 with 2 mol L-1 aqueous HCl. After stirring in the dark overnight (~15 h) at ambient temperature, the suspension was filtered. The solid product was rinsed with water, and then dried under vacuum at ambient temperature.
2.3. Modification with Al2O3
Al2O3 was deposited on Pt/TiO2 by a sol-gel process [30]. Pt/TiO2 (200 mg) was suspended in 40 mL of ethanol that contained 200 µL of aqueous 0.1 mol L-1 sulfuric acid and aluminum isopropoxide (≥ 98.0%, TCI) with a nominal amount 2 wt% Al. The suspension was sonicated for 30 min and then stirred for 1 day. The powdered product was separated by filtration, rinsed with water, and then dried under vacuum at ambient temperature.
2.4. Modification with polymer
Polymer modification was performed as previously reported [25,26]. 100 mg of dye-loaded solid material was suspended in a solution of the anionic polymer dissolved in water (0.25 mmol L-1, 40 mL), whose pH was set to be 2 by addition of HCl solution (2 mol L-1). The suspension was then stirred in the dark at ambient temperature for 1 h. The solid was then separated by filtration, rinsed with water, and dried under vacuum at ambient temperature. The used polymers were poly(sodium 4-styrenesulfonate) (Sigma-Aldrich, MW 70000), and sodium polymethacrylic acid (Sigma-Aldrich, MW 9500).
2.5. Characterization
Ultraviolet-visible (UV-vis) spectra were obtained in both diffuse reflectance (DR) and transmission modes using a spectrophotometer (V-770 and V-565, JASCO). X-ray photoelectron spectra (XPS) were obtained using an ESCA-3400 (Shimadzu). XPS binding energies for each sample were referenced to the C 1s peak (284.5 eV). A JASCO FT/IR-4600 apparatus was used to take Fourier transform infrared (FT-IR) spectra in the attenuated total reflection method with a diamond prism. A Hamamatsu Quantaurus-QY Plus apparatus was employed to measure emission quantum yields of the dye-sensitized solids. A HORIBA SZ-100 was used to measure ζ potentials. The JEM-2010F electron microscope (JEOL) in the Open Facility Center, Materials Analysis Division, Tokyo Institute of Technology was used to obtain transmission electron microscopy (TEM) images.
2.6. Photocatalysis
The procedure for studying photocatalytic reactions was the same as reported in previous papers [25,26,30]. The dye-sensitized photocatalyst sample (20 mg) was suspended in a top irradiation-type cell that contained 100 mL of an aqueous solution of the electron donor. The electron donors used were ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA·2Na, 99.5%, Dojindo Molecular Technologies) and NaI (99.5%, Kanto Chemical Co.). To adjust the reaction pH to be 4.0, aqueous sulfuric acid solution was added in the NaI reaction solution. For overall water splitting in a Z-scheme, 50 mg of PtOx/H-Cs-WO3 photocatalyst prepared by the previous literature [31] was employed as the O2-evolving photocatalyst. A 300 W xenon lamp (Cermax, PE300BF) with a CM-1 cold mirror and an L42 cutoff filter was used as the source of visible light (λ > 400 nm). A HAL-320 solar simulator was used as the light source for quantifying the solar-to-hydrogen energy conversion efficiency. The product gases were analyzed by using a Shimadzu GC-8 A gas chromatograph equipped with a TCD detector and argon carrier gas. The MS-5A column of the GC was directly connected to a glass closed gas circulation system made of glass.
The STH conversion efficiency for Z-scheme water splitting was estimated from Eq. (1):
Here RH and RO are the rates of H2 and O2 evolution (mol s- 1), respectively. ΔG° is the standard free energy of formation of liquid H2O (237 × 103 J mol-1), and P and S are the power density of simulated sunlight (100 mW cm-2), and the area of illumination (9 cm2), respectively.
The apparent quantum yield (AQY) of H2 evolution was measured using the same apparatus with a band-pass filter (λ = 420 nm), and was estimated from Eq. (2)
Here A, R, and I are the reaction coefficient, the rate of H2 evolution, and the power of incident photons, respectively. The incident photon power was found to be 43.1 mW by using a calibrated silicon photodiode.
2.7. Transient absorption measurements
An enVISion transient UV-visible spectrometer (Magnitude Instruments, State College, PA) was used to perform transient absorption spectroscopy measurements [19,25⇓-27]. Briefly, the sensitized solid (10 mg) was suspended either in water or in an aqueous solution of NaI (10 or 100 mmol L-1, 4 mL), and transient diffuse reflectance traces were obtained at selected wavelengths on timescales of nanoseconds to milliseconds following 532 nm laser excitation. The pH of the NaI solution was adjusted to ~4.0 by addition of aqueous sulfuric acid. Before the measurements, Ar purging (20 min) was made for the suspension.
3. Results and discussion
3.1. Preparation and characterization
3.1.1. Pt/TiO2
Pt was loaded onto TiO2 by a photodeposition method. TEM images of Pt-loaded TiO2 are shown in Fig. 1. Because of the electron density contrast between Pt and TiO2, the loaded Pt on TiO2 can be observed as darker spots. These images confirm that not all particles have Pt on them, and the diameter of the Pt nanoparticles was about 2-4 nm, which is similar to that reported previously [32]. Diffuse reflectance spectra also confirmed the deposition of Pt on TiO2. After Pt deposition, the metallic luster of surface Pt was readily observable (Fig. S1).
Fig. 1.

Fig. 1.
TEM images of Pt/TiO2 and Al2O3/Pt/TiO2.
3.1.2. Modified RuP/Pt/TiO2
RuP was adsorbed onto Pt/TiO2 and Al2O3-modified Pt/TiO2 (Al2O3/Pt/TiO2). The existence of Al2O3 on TiO2 was confirmed by XPS measurements (Fig. S2(a)). The Al/Ti atomic ratio in RuP/Al2O3/Pt/TiO2 was measured to be 0.2 by XPS. The deposition of Al2O3 on Pt/TiO2 was observed as an amorphous overlayer and nanoparticles (~1 nm) by TEM (Fig. 1). The morphological character of the deposited Al2O3 was similar to that reported previously [20]. Fig. S3 shows DRS of Pt/TiO2 and Al2O3/Pt/TiO2 before and after RuP adsorption. In the DRS, an absorption band derived from the singlet metal-to-ligand charge transfer (1MLCT) excitation of RuP appears at around 460 nm.
Fig. 2 shows steady-state emission spectra of RuP/Al2O3, RuP/Pt/TiO2 and RuP/Al2O3/Pt/TiO2. A strong emission peak is observed at 670 nm for RuP/Al2O3. The calculated emission quantum yield was 6.0%, close to that reported previously (4.1%) [26,30]. This emission was largely quenched on RuP-sensitized Pt/TiO2 or Al2O3/Pt/TiO2 by rapid electron transfer from the excited RuP molecule to TiO2. The emission quantum yields of RuP/Pt/TiO2 and RuP/Al2O3/Pt/TiO2 were 0.2% and 1.4%, respectively. When HCa2Nb3O10 was used instead of TiO2, the quantum yield for the red emission was 0.2% with or without Al2O3 modification [30]. These results indicate that Al2O3 modification slows the electron transfer reaction between the dye and the semiconductor when TiO2 was used, unlike HCa2Nb3O10. However, even with the Al2O3 modification, it is clear that oxidative electron injection occurs.
Fig. 2.

Fig. 2.
Steady-state emission spectra of RuP-sensitized Pt/TiO2 with various modifications. The samples were suspended in H2O (pH adjusted 4 by aqueous sulfuric acid) saturated with Ar.
FT-IR spectroscopy identified the presence of the anionic polymers on the dye-adsorbed TiO2 samples (Fig. 3). In the spectrum of the PMA-modified sample, new peaks at 1900-1600 and 1500-1000 cm-1, attributed to the stretching vibrations of the carbonyl group C=O of PMA and antisymmetric/symmetric stretching vibration of COO- of PMA [33], appeared. PSS-grafted samples exhibit new peaks at 1000-1300 cm-1, assigned to the antisymmetric and symmetric stretching vibrations of SO3- or in-plane skeletal vibrations and in-plane bending vibrations of the phenyl rings of PSS [34]. Additionally, in the case of PSS modification, a S 2p photoelectron signal at 168.0 eV, attributable to PSS was observed by XPS measurements (Fig. S2 (b)) [25]. The S/Ti atomic ratio in PSS/RuP/Pt/TiO2 was measured to be 0.07 by XPS. However, TEM observation could not identify the presence of PSS.
Fig. 3.
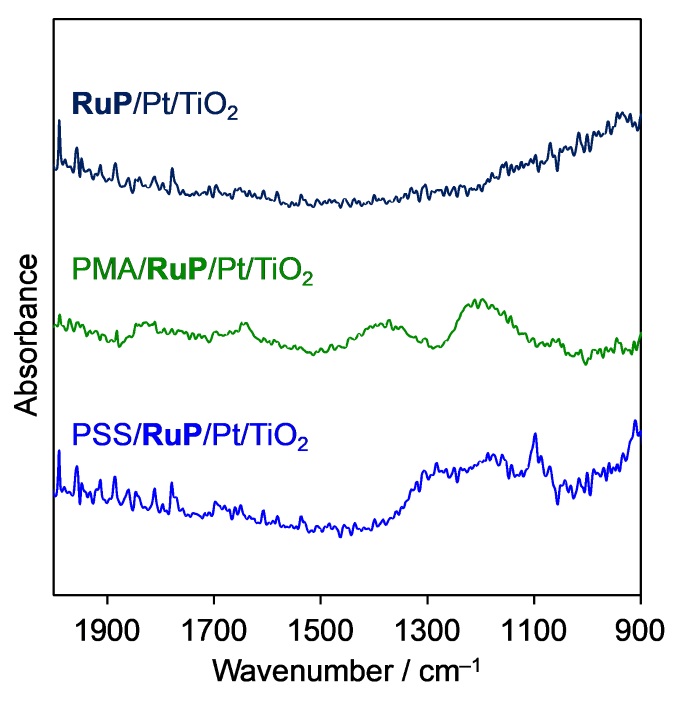
Fig. 3.
FT-IR spectra of polymer-modified RuP/Pt/TiO2.
It is also noted that the 1MLCT absorption band of RuP was redshifted by polymer modification (Fig. S4). This is the same phenomenon was observed in our previous report [26], suggesting that there is some interaction between RuP and the anionic polymer. The emission quantum yields of PMA/RuP/Pt/TiO2 and PSS/RuP/Pt/TiO2 were 0.4% and 0.1%, respectively (Fig. 2). These yields are almost identical to those of the unmodified samples (0.2%).
Fig. 4 displays the ζ-potentials of RuP/Pt/TiO2 samples with and without polymer modification. The polymer modified samples had more negative ζ-potentials, compared to the unmodified one. This result is consistent with previous reports [26]. Suppression of the back electron transfer reaction to I3- by anionic polymers originates from the repulsive Coulombic interaction between the negatively charged polymers and I3- [24⇓-26]. Therefore, at pH 4, the back electron transfer suppression effect of anionic polymers can be expected in the TiO2 case.
Fig. 4.
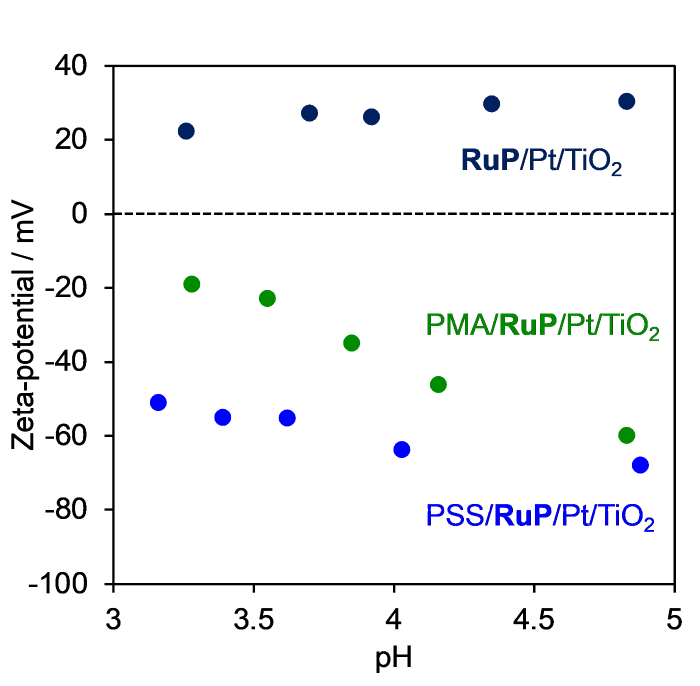
Fig. 4.
ζ-potentials of polymer-modified RuP/Pt/TiO2 as a function of pH.
3.2. Photocatalytic activity
3.2.1. H2 evolution half reaction
Fig. 5 shows the amount of H2 evolved versus time for the half reaction in 10 mmol L-1 NaI solution. Samples without any polymer or Al2O3 modification produced very little H2 in NaI solution, despite the high H2-evolution activity in aqueous EDTA solution (Fig. S5). Even when TiO2 was modified with Al2O3, the activity remained low. While the previously reported RuP/Pt(in)/HCa2Nb3O10 without Al2O3 and/or polymer modification showed lower activity than RuP/Pt/TiO2 in aqueous EDTA solution (Fig. S5), it exhibited higher H2-producing activity (Figs. 5 and S6). The positive effect of Al2O3 on H2 evolution by dye-sensitized Pt/TiO2 has been reported previously [20]. The loading amounts of Pt and RuP for TiO2 were optimized to be 0.1 wt% and 15 µmol g-1, respectively (Fig. S7).
Fig. 5.
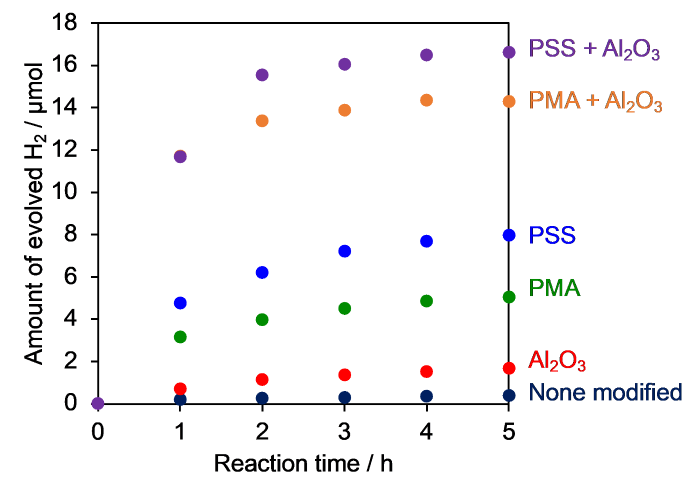
Fig. 5.
H2 evolution time courses from an aqueous NaI solution (5 mmo l L‒1) over modified RuP/Pt/TiO2 (λ > 400 nm).
The different trend of H2 evolution activity with respect to electron donor is probably due to the position of the cocatalyst. In RuP/Pt/TiO2, Pt was loaded onto the external surface of TiO2 (Fig. 1), whereas Pt was intercalated into the interlayer of HCa2Nb3O10 [30]. Since the intercalation of I3- into the Ca2Nb3O10- sheets is inhibited by electrostatic repulsion, the thermodynamically favorable Pt-catalyzed reaction between H2 and I3- can be effectively suppressed [6]. On the other hand, Pt on TiO2 has nothing to suppress the approach of I3-, so scavenging of H2 by I3- may occur efficiently even in the dark, leading to lower net H2-evolution activity. Surprisingly, this was not the case, because a dark reaction test using a Pt/TiO2 suspension containing I3- in the presence of H2 gas showed no change in the amount of H2 in the gas phase.
A clear enhancement of H2 evolution activity of RuP/Pt/TiO2 was observed upon PSS or PMA modification (Fig. 5). This result suggests that the anionic polymer can prevent I3- from accessing the TiO2 surface and Pt, thereby suppressing the catalytic oxidation of H2. This idea is supported by the result of the ζ-potential measurements (Fig. 4). The H2-evolution activity was further enhanced by modifying with both Al2O3 and the anionic polymer. This is because Al2O3 was able to suppress the back electron transfer from the semiconductor to the oxidized dye [20,27].
3.2.2. Overall water splitting according to Z-scheme
Time courses of H2 and O2 evolution for overall water-splitting in the Z-scheme are shown in Fig. 6. The Z-scheme system using the unmodified RuP/Pt/TiO2 was able to evolve both H2 and O2. Here, the PtOx/H-Cs-WO3 photocatalyst efficiently consumes the generated I3- to produce O2 in the Z-scheme [31], and the back electron transfer reactions of I3- are less likely to occur. Even for the Z-scheme, the activity was improved by anionic polymer modification (Fig. 6(a)). Al2O3 modification also resulted in an improvement in photocatalytic activity, and further improvement was observed by modifying with both anionic polymer and Al2O3 (Fig. 6(b)). In all cases, however, the ratio of H2 to O2 evolved was larger than expected from stoichiometric water splitting (i.e., H2/O2 = 2). In this Z-scheme, H2 evolution is initiated by oxidation of I- on the RuP/Pt/TiO2 photocatalyst before O2 is evolved over the PtOx/H-Cs-WO3. Therefore, some H2 was produced before a sufficient concentration of I3- was available for PtOx/H-Cs-WO3 to work steadily. The STH values of samples modified with both anionic polymer and Al2O3 were 0.016% for PMA and 0.023% for PSS, respectively (Fig. S8). These values are 5-7 times lower than those achieved by using RuP/Pt(in)/HCa2Nb3O10 with Al2O3 and PSS (or PMA) modifications [25,26]. The AQYs of RuP/Al2O3/Pt/TiO2 modified with PMA and PSS were measured to be 0.20% and 0.77% at 420 nm, respectively.
Fig. 6.

Fig. 6.
Time courses of Z-scheme overall water splitting (λ > 400 nm) from an aqueous NaI solution over polymer modified RuP/Pt/TiO2 (a) and RuP/Al2O3/Pt/TiO2 (b).
The initial rates of water splitting in the Z-scheme using unmodified RuP/Pt(in)/HCa2Nb3O10 (Fig. S9) were lower than those recorded with polymer-modified RuP/Al2O3/Pt/TiO2. When Al2O3 was loaded onto HCa2Nb3O10 to suppress the semiconductor-to-dye back electron transfer, the activity became higher than that of the fully-modified TiO2; the AQY of PSS/RuP/Al2O3/Pt/TiO2 (0.77% at 420 nm) was lower than that of RuP/Al2O3/Pt(in)/HCa2Nb3O10 (2.4%) [30]. This suggests that HCa2Nb3O10, in which the back electron transfer reaction to I3- is more effectively inhibited, is more suitable for Z-scheme water splitting using reversible electron donors.
We have reported that degradation or desorption of RuP from Pt(in)/HCa2Nb3O10 occurs during the reaction for ~5 h under high-intensity Xe lamp illumination (λ > 400 nm), resulting in a decrease in activity [30]. A similar phenomenon was observed in the present study of RuP/Pt/TiO2, because the loss of 1MLCT absorption intensity and its red-shift, indicative of the desorption and degradation of RuP, were seen in the UV visible DR spectra of RuP/Pt/TiO2 before and after the H2 evolution reaction (Fig. S10).
3.3. Transient absorption measurements
To quantify the electron transfer kinetics in RuP/Pt/TiO2, flash photolysis/transient absorption experiments were performed. It is possible to distinguish the two types of back electron transfer by observing the kinetics at two different wavelengths. Charge recombination between TiO2 and oxidized RuP was assessed by observing the change in absorption of RuP at 475 nm in water in the absence of both I- and I3- [27,30]. On the other hand, the charge transfer reaction between the TiO2 conduction band and I3- could be monitored by measuring the change in absorbance of I3- at 380 nm under conditions (100 mmol L‒1 NaI) where the oxidized dye was reduced very rapidly [24,25].
First, we compared the rates of charge recombination between the semiconductor and oxidized RuP for RuP/Pt/TiO2 and RuP/Pt(in)/HCa2Nb3O10. This rate can be measured from bleaching recovery rate of the dye in the absence of reducing agents [25,27]. Fig. 7 shows the bleaching recovery kinetics of RuP on Pt/TiO2 and Pt(in)/HCa2Nb3O10 in water, observed at 475 nm. The complex kinetics of the charge recombination reaction were modeled as a quadruple-exponential decay (Eq. (3)) with some modification from previous studies [19,27].
Fig. 7.
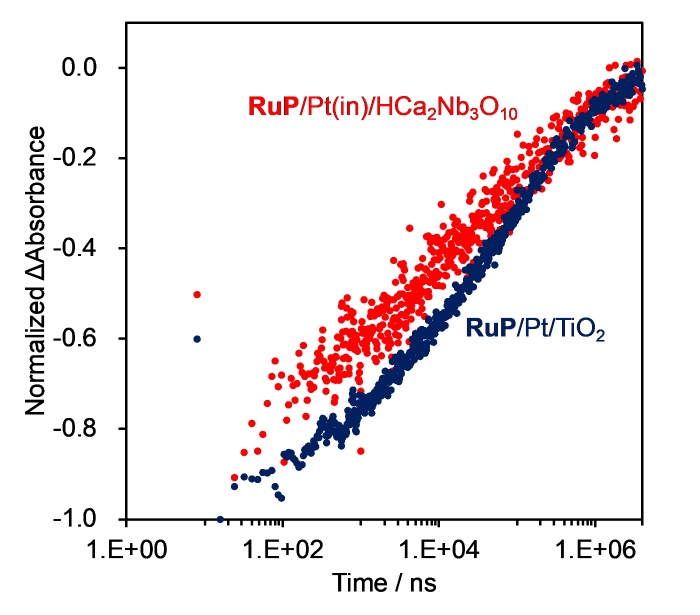
Fig. 7.
Normalized Δabsorbance at 475 nm of RuP-sensitized photocatalysts as a function of time. Experimental conditions: water (adjusted to pH 4.0 by addition of sulfuric aid); excitation wavelength, 532 nm.
The calculated lifetimes are given in Table 1. We note that the accuracy of the shortest lifetime component (τ1) is limited by convolution with the timescale of laser excitation (16 ns). Therefore, only τ2-τ4 are discussed here. The fastest kinetic component (τ2) can be attributed to rapid charge recombination between the semiconductor and oxidized RuP. The middle-long lifetime component (τ3) is assigned to back electron transfer from defect levels in the semiconductor to RuP [19,27]. The slowest component (τ4) is attributed to charge recombination by oxidized RuP molecules that are well-separated from electrons in the conduction band of the semiconductor, electrons in deep defect levels, and/or electrons at the cocatalyst [19,27]. For all lifetime components, TiO2 has a longer lifetime than HCa2Nb3O10. This can be explained by the relative conduction band potentials of the two semiconductors. The conduction band edge potential of TiO2 is at least 0.3 V more positive than that of HCa2Nb3O10 [30,35]. This means that the difference between the potential of RuP (RuP3+/RuP2+) and the conduction band of the semiconductor is larger in HCa2Nb3O10. Therefore, this could be one of the reasons why the lifetime of reverse electron transfer from semiconductor to dye is longer in TiO2. The longer lifetime components of RuP/Pt/TiO2 relative to RuP/Pt(in)/HCa2Nb3O10 (in other words, slower back electron transfer reactions) could explain the higher H2 production activity from aqueous EDTA solutions, where reduction of the oxidized sensitizer competes kinetically with back electron transfer (Fig. S5).
Table 1 Back electron transfer kinetics of RuP adsorbed on Pt/TiO2 or Pt(in)/HCa2Nb3O10 in water a.
| Catalyst | Absorption decay lifetimes / µs (%) | |||
|---|---|---|---|---|
| τ1 | τ2 | τ3 | τ4 | |
| RuP/Pt/TiO2 | 0.1 | 3.5 ± 0.4 | 50 ± 7 | 500 ± 60 |
| (30) | (35) | (35) | ||
| RuP/Pt(in)/HCa2Nb3O10 | 0.09 | 2.0 ± 0.5 | 20 ± 4 | 250 ± 50 |
| (27) | (29) | (44) | ||
a Experimental conditions: water (adjusted to pH 4.0 by addition of sulfuric acid); excitation wavelength, 532 nm; monitoring wavelength, 475 nm.
In addition, measurements were also performed using TiO2 modified with polymers or Al2O3 (Fig. S11). The bleaching recovery was not changed by the polymer modification. However, the Al2O3-modified sample showed a slower bleaching recovery. This indicates that charge recombination between the semiconductor to the dye is suppressed by Al2O3. These results are consistent with those reported using HCa2Nb3O10 [25].
It is possible to quantify the reactivity of electrons in the semiconductor conduction band with the I3- ion by measuring the change in the 380 nm transient absorbance, where I3- absorbs strongly [24⇓-26]. Fig. 8 shows the time courses of transient absorbance recorded at 380 nm, and Table 2 summarizes the measured lifetimes. The kinetics were fit to either double- or triple-exponential functions (Eqs. (4) and (5)).
Fig. 8.
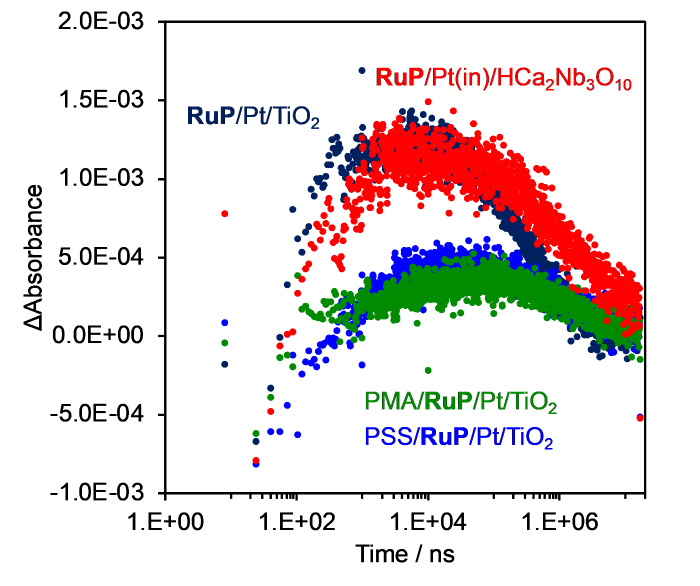
Fig. 8.
Normalized Δabsorbance at 380 nm of RuP-sensitized photocatalysts as a function of time. Measurement conditions: aqueous 100 mmol L‒1 NaI solution (adjusted to pH 4.0 by addition of sulfuric acid); excitation wavelength, 532 nm.
Table 2 Absorption decay lifetimes of I3-.
| Catalyst | Absorption decay lifetimes / µs (%) | ||
|---|---|---|---|
| τ1 | τ2 | τ3 | |
| RuP/Pt/TiO2 | 0.2 ± 0.02 | 150 ± 30 | 900 ± 200 |
| (48) | (31) | (21) | |
| RuP/Pt(in)/HCa2Nb3O10 | 0.5 ± 0.05 | 400 ± 140 | 3000 ± 1000 |
| (55) | (17) | (28) | |
| PMA/RuP/Pt/TiO2 | 5.0 ± 0.9 | 1500 ± 200 | - |
| (43) | (57) | - | |
| PSS/RuP/Pt/TiO2 | 3.0 ± 0.3 | 500 ± 100 | 2000 ± 400 |
| (52) | (24) | (24) | |
For all these samples, there was an initial increase (τ1) and subsequent decay (τ2, τ3) of ΔAbs., attributable to an initial fast increase and slower decrease in I3- concentration, respectively. First, let us consider the increase of ΔAbs., which is faster for TiO2 than for HCa2Nb3O10. This means that the reaction between oxidized RuP and I- is faster on TiO2, which is consistent with the measured recovery of RuP absorption in a 10 mmol L-1 NaI solution (Fig. S12) [25,26]. This difference corresponds to the difference in ζ-potentials between TiO2 and HCa2Nb3O10. At pH 4.0, the ζ-potential of TiO2 is positive (Fig. 4), whereas that of HCa2Nb3O10 is rather negative [25,30]. Therefore, TiO2 may attract I- more readily so that the concentration of I3- increases more rapidly upon photoexcitation. This idea is supported by the fact that the τ1 of RuP/Pt/TiO2 became much longer and the absorption change (ΔAbs.) smaller upon anionic polymer modification (PMA or PSS), which caused a large negative shift in the ζ-potential (Fig. 4).
τ2 and τ3 correspond to the kinetics of back electron transfer between I3- and the photocatalyst. The appearance of two components is presumably associated with the reactions at the Pt cocatalyst and the TiO2 surface. In the case of RuP/Pt/TiO2, both τ2 and τ3 are longer after modification with PMA or PSS. This is attributed to the Coulombic repulsion between the negatively charged surface of the photocatalyst and I3- ions, consistent with previous reports [24⇓-26]. However, despite the anionic polymer modification, PMA/RuP/Pt/TiO2 and PSS/RuP/Pt/TiO2 had shorter τ2 and τ3 values than RuP/Pt(in)/HCa2Nb3O10. Therefore, the anionic polymer-modified RuP/Pt/TiO2 can react with I3- more effectively. This is consistent with its lower H2 evolution activity relative to RuP/Pt(in)/HCa2Nb3O10 (Figs. 5 and S8).
4. Conclusions
RuP-sensitized, Pt-loaded TiO2 was studied as a photocatalyst for visible light H2 evolution in the presence of EDTA or NaI as electron donors. Compared to a similar dye-sensitized photocatalyst (RuP/Pt(in)/HCa2Nb3O10), RuP/Pt/TiO2 showed higher H2 evolution activity from aqueous EDTA solution, but lower H2 yield from NaI solution. Although modification of RuP/Pt/TiO2 with anionic polymers (e.g., PMA or PSS) improved the H2 evolution activity from aqueous NaI, the modified materials did not outperform unmodified RuP/Pt(in)/HCa2Nb3O10. In Z-scheme overall water splitting with PtOx/H-Cs-WO3, the activity of RuP/Pt/TiO2 even with the optimal surface modification was much lower than that of the RuP/Pt(in)/HCa2Nb3O10 counterpart. Transient absorption measurements indicated that charge recombination between the semiconductor and the dye occurs less readily in RuP/Pt/TiO2 than in RuP/Pt(in)/HCa2Nb3O10, but back electron transfer reactions with I3- are more rapid on RuP/Pt/TiO2. Although the back electron transfer reaction with I3- could be suppressed by modifying anionic polymers (PMA or PSS) to a certain extent, Pt(in)/HCa2Nb3O10 possessing intrinsically negative surface charge exhibited a more pronounced suppression effect. Based on these results, it can be concluded that Pt/TiO2 is useful when sacrificial reducing agents such as EDTA are used. However, with a reversible electron donor such as I-, it is necessary to remodel the photocatalyst to inhibit the reaction with an electron acceptor, which directly lowers the forward electron transfer reaction. Improvement of the forward electron transfer yield is possible by refining the cocatalyst employed in the dye-sensitized H2 evolution system [19], as a good cocatalyst facilitates the extraction of electrons from the semiconductor and the reduction of H+ (or H2O) [36,37].
Acknowledgments
K.M. acknowledge support from JSPS-KAKENHI (P22H01862, JP22H05142, JP22H05148, JPJSCCA20200004). T.E.M. and L.X. thank support from the US Department of Energy (DE-SC0019781).
Electronic supporting information
Supporting information is available in the online version of this article.
Reference

One-directional electron transport between a photocatalyst and redox mediator is crucial for achieving highly active Z-scheme water-splitting photocatalysis. Herein, a photoredox cascade catalyst that artificially mimics the electron transport chain in natural photosynthesis was synthesized from a Pt-TiO nanoparticle catalyst, two photosensitizers ( and ), and a visible-light-transparent electron mediator (). During photocatalytic hydrogen evolution in the presence of a redox-reversible electron donor, [Co(bpy)] (bpy = 2,2'-bipyridine), the -Zr--Zr-@Pt-TiO () photocatalyst exhibited the highest reported initial (1 h) apparent quantum yield (AQY = 2.23%) of dye-sensitized TiO photocatalysts to date. Furthermore, successfully produced hydrogen when using hydroquinone monosulfonate (HQS) as the hydrogen source.
Conversion of sunlight into chemical energy has been the subject of intense research efforts in recent years, due to the possibility to store the enormous amount of energy continuously provided by the Sun in the form of useful "solar fuels". To allow such process, suitable photocatalysts are required, which are usually obtained by combination of different inorganic materials or by self-assembly of molecular components on semiconductor surfaces: accordingly, they are capable to carry out several different processes such as light harvesting, substrate binding, electron transport, storage and transfer. Among the photocatalytic systems developed to date, heterogeneous dye-sensitized semiconductors decorated with various electrocatalysts received particular attention, thanks to their favorable properties of tunable absorption spectrum, high stability and adaptability to different reactions. In this paper, we will review some selected examples of application of such photocatalysts to two main reactions, namely H+ reduction to H-2 and CO2 conversion to C-1-building blocks (CO, formate), which are among the most important transformations in the field of so-called "artificial photosynthesis".
We report herein a methodology for conformally coating nanocrystalline TiO2 films with a thin overlayer of a second metal oxide. SiO2, Al2O3, and ZrO2 overlayers were fabricated by dipping mesoporous, nanocrystalline TiO2 films in organic solutions of their respective alkoxides, followed by sintering at 435 degrees C. These three metal oxide overlayers are shown in all cases to act as barrier layers for interfacial electron transfer processes. However, experimental measurements of film electron density and interfacial charge recombination dynamics under applied negative bias were vary significantly for the overlayers. A good correlation was observed between these observations and the point of zero charge of the different metal oxides. On this basis, it is found that the most basic overlayer coating, Al2O3 (pzc = 9.2), is optimal for retarding interfacial recombination losses under negative applied bias. These observations show good correlation with current/voltage analyses of dye sensitized solar cell fabricated from these films, with the Al2O3 resulting in an increase in V(oc) of up to 50 mV and a 35% improvement in overall device efficiency. These observations are discussed and compared with an alternative TiCl4 posttreatment of nanocrystalline TiO2 films with regard to optimizing device efficiency.
In the design of light-harvesting chromophores for use in dye-sensitized photoelectrosynthesis cells (DSPECs), surface binding to metal oxides in aqueous solutions is often inhibited by synthetic difficulties. We report here a systematic synthesis approach for preparing a family of Ru(II) polypyridyl complexes of the type [Ru(4,4'-R2-bpy)2(4,4'-(PO3H2)2-bpy)](2+) (4,4'(PO3H2)2-bpy = [2,2'-bipyridine]-4,4'-diylbis(phosphonic acid); 4,4'-R2-bpy = 4,4'-R2-2,2'-bipyridine; and R = OCH3, CH3, H, or Br). In this series, the nature of the 4,4'-R2-bpy ligand is modified through the incorporation of electron-donating (R = OCH3 or CH3) or electron-withdrawing (R = Br) functionalities to tune redox potentials and excited-state energies. Electrochemical measurements show that the ground-state potentials, E(o')(Ru(3+/2+)), vary from 1.08 to 1.45 V (vs NHE) when the complexes are immobilized on TiO2 electrodes in aqueous HClO4 (0.1 M) as a result of increased Ru dπ-π* back-bonding caused by the lowering of the π* orbitals on the 4,4'-R2-bpy ligand. The same ligand variations cause a negligible shift in the metal-to-ligand charge-transfer absorption energies. Emission energies decrease from λmax = 644 to 708 nm across the series. Excited-state redox potentials are derived from single-mode Franck-Condon analyses of room-temperature emission spectra and are discussed in the context of DSPEC applications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}